
Mass spectrometry (also known as mass spectroscopy (deprecated)or informally, "mass-spec" and MS) is an analytical technique used to measure the mass-to-charge ratio of ions. It is most generally used to find the composition of a physical sample by generating a mass spectrum representing the masses of sample components. The mass spectrum is measured by a mass spectrometer.
All mass spectrometers consist of three basic parts: an ion source, a mass analyzer, and a detector system. The stages within the mass spectrometer are:
- Producing ions from the sample
- Separating ions of differing masses
- Detecting the number of ions of each mass produced
- Collating the data and generating the mass spectrum
The technique has several applications, including;

- identifying unknown compounds by the mass of the compound molecules or their fragments
- determining the isotopic composition of elements in a compound
- determining the structure of a compound by observing its fragmentation
- quantifying the amount of a compound in a sample using carefully designed methods (mass spectrometry is not inherently quantitative)
- studying the fundamentals of gas phase ion chemistry (the chemistry of ions and neutrals in vacuum)
- determining other physical, chemical, or even biological properties of compounds with a variety of other approaches
Instrumentation
Ion source
The ion source is the part of the mass spectrometer that ionizes the material under analysis (the analyte). The ions are then transported by magnetic or electric fields to the mass analyzer.
Techniques for ionization have been key to determining what types of samples can be analyzed by mass spectrometry. Electron ionization and chemical ionization are used for gases and vapors. In chemical ionization sources, the analyte is ionized by chemical ion-molecule reactions during collisions in the source. Two techniques often used with liquid and solid biological samples include electrospray ionization (due to John Fenn) and matrix-assisted laser desorption/ionization (MALDI, due to K. Tanaka and separately, M. Karas and F. Hillenkamp). Inductively coupled plasma sources are used primarily for metal analysis on a wide array of sample types. Others include glow discharge, fast atom bombardment (FAB), thermospray, desorption/ionization on silicon (DIOS), Direct Analysis in Real Time (DART), atmospheric pressure chemical ionization (APCI), secondary ion mass spectrometry (SIMS), spark ionization and thermal ionisation.
Mass analyzer
Mass analyzers separate the ions according to their mass-to-charge ratio. All mass spectrometers are based on dynamics of charged particles in electric and magnetic fields in vacuum where the following two laws apply:
 (
(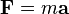 (Newton's second law of motion)
(Newton's second law of motion)
where F is the force applied to the ion, m is the mass of the ion, a is the acceleration, q is the ionic charge, E is the electric field, and v x B is the vector cross product of the ion velocity and the magnetic field
Equating the above expressions for the force applied to the ion yields:

This differential equation is the classic equation of motion of charged particles. Together with the particle's initial conditions it completely determines the particle's motion in space and time and therefore is the basis of every mass spectrometer. It immediately reveals that two particles with the same physical quantity m/q behave exactly the same. Thus all mass spectrometers actually measure m/q and strictly speaking should be called mass-to-charge spectrometers. When presenting data, it is common to use the (officially) dimensionless m/z (called mass-to-charge ratio, although (more accurately) it represents the ratio of the mass number and the charge number), where z is the number of elementary charges (e) on the ion (z=q/e).
There are many types of mass analyzers, using either static or dynamic fields, and magnetic or electric fields, but all operate according to this same law. Each analyzer type has its strengths and weaknesses. Many mass spectrometers use two or more mass analyzers for tandem mass spectrometry (MS/MS). In addition to the more common mass analyzers listed below, there are other less common ones designed for special situations.
Sector
A sector field mass analyzer uses an electric and/or magnetic field to affect the path and/or velocity of the charged particles in some way. As shown above, sector instruments change the direction of ions that are accelerated through the mass analyzer. The ions enter a magnetic or electric field which bends the ion paths depending on their mass-to-charge ratios, deflecting the more charged and faster-moving, lighter ions more. The ions eventually reach the detector and their relative abundances are measured. The analyzer can be used to select a narrow range of m/q or to scan through a range of m/q to catalog the ions present.
Time-of-flight
Perhaps the easiest to understand is the Time-of-flight (TOF) analyzer. It uses an electric field to accelerate the ions through the same potential, and then measures the time they take to reach the detector. If the particles all have the same charge, then their kinetic energies will be identical, and their velocities will depend only on their masses. Lighter ions will reach the detector first.
Quadrupole .
Quadrupole mass analyzers use oscillating electrical fields to selectively stabilize or destabilize ions passing through a radio frequency (RF) quadrupole field. A quadrupole mass analyzer acts as a mass selective filter and is closely related to the Quadrupole ion trap, particularly the linear quadrupole ion trap except that it operates without trapping the ions. A common variation of the quadrupole is the triple quadrupole.
Quadrupole ion trap.
The quadrupole ion trap works on the same physical principles as the QMS, but the ions are trapped and sequentially ejected. Ions are created and trapped in a mainly quadrupole RF potential and separated by m/q, non-destructively or destructively.
There are many mass/charge separation and isolation methods but most commonly used is the mass instability mode in which the RF potential is ramped so that the orbit of ions with a mass a > b are stable while ions with mass b become unstable and are ejected on the z-axis onto a detector.
Ions may also be ejected by the resonance excitation method, whereby a supplemental oscillatory excitation voltage is applied to the endcap electrodes, and the trapping voltage amplitude and/or excitation voltage frequency is varied to bring ions into a resonance condition in order of their mass/charge ratio.
The cylindrical ion trap mass spectrometer is a derivative of the quadrupole ion trap mass spectrometer.
Linear quadrupole ion trap
A linear quadrupole ion trap (LTQ) is similar to a QIT, but traps ions in a 2D quadrupole field, instead of a 3D quadrupole field as in a QIT. Ions can be stored along the entire length of the LTQ which results in a higher ion capacity.
Fourier transform ion cyclotron resonance .
Fourier transform mass spectrometry, or more precisely Fourier transform ion cyclotron resonance MS, measures mass by detecting the image current produced by ions cyclotroning in the presence of a magnetic field. Instead of measuring the deflection of ions with a detector such as an electron multiplier, the ions are injected into a Penning trap (a static electric/magnetic ion trap) where they effectively form part of a circuit. Detectors at fixed positions in space measure the electrical signal of ions which pass near them over time producing cyclical signal. Since the frequency of an ion's cycling is determined by its mass to charge ratio, this can be deconvoluted by performing a Fourier transform on the signal. FTMS has the advantage of high sensitivity (since each ion is 'counted' more than once) and much high resolution and thus precision.[8][9]
Ion cyclotron resonance is an older mass analysis technique similar to FTMS except that ions are detected with a traditional detector. Ions trapped in a Penning trap are excited by an RF electric field until they impact the wall of the trap where the detector is located with ions of different mass being resolved in time.
Orbitrap
The Orbitrap is the most recently introduced mass analyser (commercially available since 2005,ThermoElectron(R)). In the Orbitrap, ions are electrostatically trapped in an orbit around a central, spindle-shaped electrode. The electrode confines the ions so that they both orbit around the central electrode and oscillate back and forth along the central electrode's long axis. This oscillation generates an image current in the detector plates which is recorded by the instrument. The frequencies of these image currents depend on the mass to charge ratios of the ions in the Orbitrap. Mass spectra are obtained by Fourier transformation of the recorded image currents.
Similar to Fourier transform ion cyclotron resonance mass spectrometers, Orbitraps have a high mass accuracy, high sensitivity and a good dynamic range.
Detector
The final element of the mass spectrometer is the detector. The detector records the charge induced or current produced when an ion passes by or hits a surface. In a scanning instrument the signal produced in the detector during the course of the scan versus where the instrument is in the scan (at what m/q) will produce a mass spectrum, a record of ions as a function of m/q.
Typically, some type of electron multiplier is used, though other detectors including Faraday cups and ion-to-photon detectors are also used. Because the number of ions leaving the mass analyzer at a particular instant is typically quite small, significant amplification is often necessary to get a signal. Microchannel Plate Detectors are commonly used in modern commercial instruments.[11] In FTMS and Orbitraps, the detector consists of a pair of metal surfaces within the mass analyzer/ion trap region which the ions only pass near as they oscillate. No DC current is produced, only a weak AC image current is produced in a circuit between the electrodes. Other inductive detectors have also been used.
Tandem MS (MS/MS)
Tandem mass spectrometry involves multiple steps of mass selection or analysis, usually separated by some form of fragmentation. A tandem mass spectrometer is one capable of multiple rounds of mass spectrometry. For example, one mass analyzer can isolate one peptide from many entering a mass spectrometer. A second mass analyzer then stabilizes the peptide ions while they collide with a gas, causing them to fragment by collision-induced dissociation (CID). A third mass analyzer then catalogs the fragments produced from the peptides. Tandem MS can also be done in a single mass analyzer over time as in a quadrupole ion trap. There are various methods for fragmenting molecules for tandem MS, including collision-induced dissociation (CID), electron capture dissociation (ECD), electron transfer dissociation (ETD), infrared multiphoton dissociation (IRMPD) and blackbody infrared radiative dissociation (BIRD). An important application using tandem mass spectrometry is in protein identification.
Tandem mass spectrometry enables a variety of experiments. Although it allows for many uniquely designed experiments some types of experiments are commonly used and built into many commercial mass spectrometers. Examples of these include single reaction monitoring (SRM), multiple reaction monitoring (MRM) and precursor ion scan. In single reaction monitoring the first analyzer allows only a single mass through and the second analyzer monitors for a specifically defined fragment ion. MRM is nearly identical except the second analyzer monitors multiple user defined fragment ions. These monikers are most often used with scanning instruments where the second mass analysis event is duty cycle limited. These experiments are used to increase specificity of detection of known molecules such as in pharmacokinetic studies. Precursor ion scan refers to monitoring for a specific loss from the precursor ion. The first and second mass analyzers scan across the spectrum separated by a user defined m/z value. This experiment is used to detect specific motifs within unknown molecules.
Common Mass Spectrometer Configurations & Techniques
When all of the elements (source, analyzer and detector) of a mass spectrometer are combined to form a complete instrument and the specific configuration becomes common a new name, often an abbreviation of one or more of the internal components, becomes attached to the specific configuration and can become, within certain circles, more well known than the specific internal components. The most ubiquitous example of this is MALDI-TOF, which simply refers to combining a Matrix-assisted laser desorption/ionization source with a Time-of-flight mass analyzer. The MALDI-TOF moniker is, however, often more widely recognized by the non-mass spectrometrist scientist than MALDI or TOF individually as if inseparable. Other examples include inductively coupled plasma-mass spectrometry (ICP-MS), accelerator mass spectrometry (AMS), Thermal ionization-mass spectrometry (TIMS) and spark source mass spectrometry (SSMS). Sometimes the use of the generic "MS" actually implies a very specific mass analyzer and detection system as with AMS, which is always sector based. In other cases there are common configurations that may be implied but not necessarily.
Certain applications of mass spectrometry have developed monikers that although technically referring to a broad application also tend to indicate a specific or a limited number of instrument configurations. An example of this is isotope ratio mass spectrometry (IRMS). Despite only specifically indicating an application, the use of a limited number of sector based mass analyzers is implied and the name is used to refer to both the application and the instrument used for the application.
Other Separation Techniques Combined with Mass spectrometry
An important enhancement to the mass resolving and determining capacity of mass spectrometry is the combination of mass spectrometry with analysis techniques that the resolve mixtures of compounds in a sample based on other characteristics before introduction into the mass spectrometer.
Gas chromatography/MS
A common form of mass spectrometry is gas chromatography-mass spectrometry (GC/MS or GC-MS). In this technique, a gas chromatograph is used to separate different compounds. This stream of separated compounds is fed on-line into the ion source, a metallic filament to which voltage is applied. This filament emits electrons which ionize the compounds. The ions can then further fragment, yielding predictable patterns. Intact ions and fragments pass into the mass spectrometer's analyser and are eventually detected.
Liquid chromatography/MS
Similar to gas chromatography MS (GC/MS), liquid chromatography mass spectrometry (LC/MS or LC-MS) separates compounds chromatographically before they are introduced to the ion source and mass spectrometer. It differs from GC/MS in that the mobile phase is liquid, usually a combination of water and organic solvents, instead of gas. Most commonly, an electrospray ionization source is used in LC/MS.
IMS/MS
Ion mobility spectrometry/mass spectrometry is a technique where ions are first separated by drift time through some pressure of neutral gas given an electrical potential gradient before being introduced into a mass spectrometer.
The drift time is a measure of the radius relative to the charge of the ion. The duty cycle of IMS (time over which the experiment takes place) is longer than most mass spectrometers such that the mass spectrometer can sample along the course of the IMS separation. This produces data about the IMS separation and the mass-to-charge ratio of the ions in a manner similar to LC/MS.
The duty cycle of IMS is short relative to liquid chromatography or gas chromatography separations and can thus be coupled to such techniques producing triply hyphenated techniques such as LC/IMS/MS.
Data and analysis
Data representations
Mass spectrometry produces various types of data. The most ubiquitous data representation is the mass spectrum.
Certain types of mass spectrometry data are best represented as a mass chromatogram. Types of chromatograms include selected ion monitoring (SIM), total ion current (TIC), and selected reaction monitoring chromatogram (SRM), among many others.
Other types of mass spectrometry data are well represented as a contour map of mass-to-charge on one axis, intensity on another and an additional experimental parameter (often time) on the third axis, thus producing a three dimensional surface.
Data analysis
Basics
Mass spectrometry data analysis is a complicated subject matter that is very specific to the type of experiment producing the data. There are several general subdivisions of data that are fundamental to beginning to understand any data.
Many mass spectrometers work in either negative ion mode or positive ion mode. It is very important to know whether the observed ions are negatively or positively charged. This is often important in determining the neutral mass but it also indicates something about the nature of the molecules.
There are many different types of ion sources that behave very differently from each other. A source such as an electron ionization source produces many fragments and mostly odd electron species with one charge, whereas a source such as an electrospray source usually produces quasimolecular even electron species that may be multiply charged.
Tandem mass spectrometry purposely produces fragment ions post-source and can drastically change the sort of data achieved by an experiment.
By understanding the origin of a sample certain expectations can be assumed. For example, if the sample is coming from a synthesis/manufacturing process impurities are likely to be present that are related to the major component. If the sample is a relatively crude preparation of a biological sample, the sample likely contains a certain amount of salt that may form adducts with the analyte molecules in certain analyses.
Results can also depend heavily on how was the sample prepared and how was it run/introduced. An important example is which matrix was used for MALDI spotting, since much of the energetics of the desorption/ionization event is controlled by the matrix rather than the laser power. Sometimes samples are spiked with sodium or another ion-carrying species to produce adducts rather than a protonated species.
The most commonly overlooked basic question by non-mass spectrometrists trying to use mass spectrometry or interact with a mass spectrometrist is what is the over-arching goal of the project. To interpret data one must know the desired outcome (and have collected the right data in the first place). There are many bits of information that can be gleaned from mass spectrometry data, such as the masses of the molecules, the purity of the sample, and the structure of the molecules. Each of these questions requires a different approach. Simply asking for a "mass-spec" will most likely not answer the real question at hand.
Applications
Isotope ratio MS: isotope dating and tracking

Mass spectrometry is also used to determine the isotopic composition of elements within a sample. Differences in mass among isotopes of an element are very small, and the less abundant isotopes of an element are typically very rare, so a very sensitive instrument is required. These instruments, sometimes referred to as isotope ratio mass spectrometers (IR-MS), usually use a single magnet to bend a beam of ionized particles towards a series of Faraday cups which convert particle impacts to electric current. A fast on-line analysis of deuterium content of water can be done using Flowing afterglow mass spectrometry, FA-MS. Probably the most sensitive and accurate mass spectrometer for this purpose is the accelerator mass spectrometer (AMS). Isotope ratios are important markers of a variety of processes. Some isotope ratios are used to determine the age of materials for example as in carbon dating. Labelling with stable isotopes is also used for protein quantification. (see Protein quantitation below)
Trace gas analysis
Several techniques use ions created in a dedicated ion source injected into a flow tube or a drift tube: selected ion flow tube (SIFT-MS), and proton transfer reaction (PTR-MS), are variants of chemical ionization dedicated for trace gas analysis of air, breath or liquid headspace using well defined reaction time allowing calculations of analyte concentrations from the known reaction kinetics without the need for internal standard or calibration.
Atom Probe
An atom probe is an instrument that combines time-of-flight mass spectrometry and field ion microscopy (FIM) to map the location of individual atoms.
Pharmacokinetics
Pharmacokinetics is often studied using mass spectrometry because of the complex nature of the matrix (often blood or urine) and the need for high sensitivity to observe low dose and long time point data. The most common instrumentation used in this application is LC-MS with a triple quadrupole mass spectrometer. Tandem mass spectrometry is usually employed for added specificity. Standard curves and internal standards are used for quantitation of usually a single pharmaceutical in the samples. The samples represent different time points as a pharmaceutical is administered and then metabolized or cleared from the body. Blank or t=0 samples taken before administration are important in determining background and insuring data integrity with such complex sample matrices. Much attention is paid to the linearity of the standard curve; however it is not uncommon to use curve fitting with more complex functions such as quadratics since the response of most mass spectrometers is less than linear across large concentration ranges.
There is currently considerable interest in the use of very high sensitivity mass spectrometry for microdosing studies, which are seen as a promising alternative to animal experimentation.
Mass spectrometry of proteins
Mass spectrometry is an important emerging method for the characterization of proteins. The two primary methods for ionization of whole proteins are electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). In keeping with the performance and mass range of available mass spectrometers, two approaches are used for characterizing proteins. In the first, intact proteins are ionized by either of the two techniques described above, and then introduced to a mass analyser. In the second, proteins are enzymatically digested into smaller peptides using an agent such as trypsin or pepsin. Other proteolytic digest agents are also used. The collection of peptide products are then introduced to the mass analyser. This is often referred to as the "bottom-up" approach of protein analysis.
Whole protein mass analysis is primarily conducted using either time-of-flight (TOF) MS, or Fourier transform ion cyclotron resonance (FT-ICR). These two types of instrument are preferable here because of their wide mass range, and in the case of FT-ICR, its high mass accuracy. Mass analysis of proteolytic peptides is a much more popular method of protein characterization, as cheaper instrument designs can be used for characterization. Additionally, sample preparation is easier once whole proteins have been digested into smaller peptide fragments. The most widely used instrument for peptide mass analysis is the quadrupole ion trap. Multiple stage quadrupole-time-of-flight and MALDI time-of-flight instruments also find use in this application.
Protein and peptide fractionation coupled with mass spectrometry
Proteins of interest to biological researchers are usually part of a very complex mixture of other proteins and molecules that co-exist in the biological medium. This presents two significant problems. First, the two ionization techniques used for large molecules only work well when the mixture contains roughly equal amounts of constituents, while in biological samples, different proteins tend to be present in widely differing amounts. If such a mixture is ionized using electrospray or MALDI, the more abundant species have a tendency to "drown" signals from less abundant ones. The second problem is that the mass spectrum from a complex mixture is very difficult to interpret because of the overwhelming number of mixture components. This is exacerbated by the fact that enzymatic digestion of a protein gives rise to a large number of peptide products.
To contend with this problem, two methods are widely used to fractionate proteins, or their peptide products from an enzymatic digestion. The first method fractionates whole proteins and is called two-dimensional gel electrophoresis. The second method, high performance liquid chromatography is used to fractionate peptides after enzymatic digestion. In some situations, it may be necessary to combine both of these techniques.
Gel spots identified on a 2D Gel are usually attributable to one protein. If the identity of the protein is desired, the gel spot can be excised, and digested proteolytically. The peptide masses resulting from the digestion can be determined by mass spectrometry using peptide mass fingerprinting. If this information does not allow unequivocal identification of the protein, its peptides can be subject to tandem mass spectrometry.
Characterization of protein mixtures using HPLC/MS is also called shotgun proteomics and mudpit. A peptide mixture that results from digestion of a protein mixture is fractionated by one or two steps of liquid chromatography. The eluent from the chromatography stage can be either directly introduced to the mass spectrometer through electrospray ionization, or laid down on a series of small spots for later mass analysis using MALDI.
Protein identification
There are two main ways MS is used to identify proteins. Peptide mass fingerprinting (mentioned in the previous section) uses the masses of proteolytic peptides as input to a search of a database of predicted masses that would arise from digestion of a list of known proteins. If a protein sequence in the reference list gives rise to a significant number of predicted masses that match the experimental values, there is some evidence that this protein was present in the original sample.

Tandem MS is becoming a more popular experimental method for identifying proteins. Collision-induced dissociation is used in mainstream applications to generate a set of fragments from a specific peptide ion. The fragmentation process primarily gives rise to cleavage products that break along peptide bonds. Because of this simplicity in fragmentation, it is possible to use the observed fragment masses to match with a database of predicted masses for one of many given peptide sequences. Tandem MS of whole protein ions has been investigated recently using electron capture dissociation and has demonstrated extensive sequence information in principle but is not in common practice. This is sometimes referred to as the "top-down" approach in that it involves starting with the whole mass and then pulling it apart rather than starting with pieces (proteolytic fragments) and piecing the protein back together using De novo repeat detection (bottom-up).
A number of different algorithmic approaches have been described to identify peptides and proteins from tandem mass spectrometry (MS/MS), peptide de novo sequencing and sequence tag based searching.
A popular option that combines a comprehensive range of data analysis features is PEAKS *.
Other existing mass spec analysis software include: Peptide fragment fingerprinting SEQUEST, Mascot, OMSSA and X!Tandem). Peptide de novo sequencing (LuteFisk, PepNovo, and Sherenga). Peptide sequence tag based searching (SPIDER, InsPecT, and GutenTAG).
Protein quantitation
Several recent methods allow for the quantitation of proteins by mass spectrometry. Typically, stable (e.g. non-radioactive) heavier isotopes of carbon (C13) or nitrogen (N15) are incorporated into one sample while the other one is labelled with corresponding light isotopes (e.g. C12 and N14). The two samples are mixed before the analysis. Peptides derived from the different samples can be distinguished due to their mass difference. The ratio of their peak intensities corresponds to the relative abundance ratio of the peptides (and proteins). The most popular methods for isotope labelling are SILAC (stable isotope labelling with amino acids in cell culture), trypsin-catalyzed O18 labeling, ICAT (isotope coded affinity tagging), ITRAQ (isotope tags for relative and absolute quantitation). “Semi-quantitative” mass spectrometry can be performed without labeling of samples. Typically, this is done with MALDI analysis (in linear mode). The peak intensity, or the peak area, from individual molecules (typically proteins) is here correlated to the amount of protein in the sample. However, the individual signal depends on the primary structure of the protein, on the complexity of the sample, and on the settings of the instrument.
Protein structure
Characteristics indicative of the 3 dimensional structure of proteins can be probed with mass spectrometry in various ways. By using chemical crosslinking to couple parts of the protein that are close in space, but far apart in sequence, information about the overall structure can be inferred. By following the exchange of amide protons with deuterium from the solvent, it is possible to probe the solvent accessibility of various parts of the protein.
No comments:
Post a Comment